Author: Deanna L. Kroetz, PhD, and Tore Stage, PhD, MSc Pharm on April 14, 2021 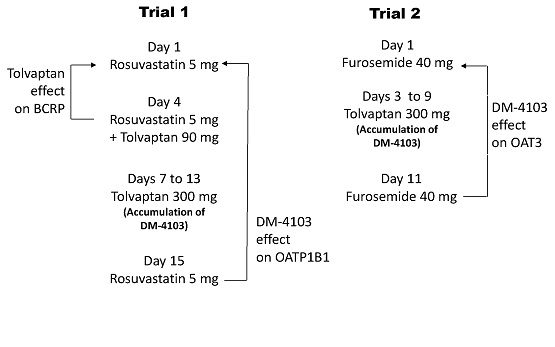
A key step during research and development of small molecule drugs is assessing absorption, distribution, metabolism, and excretion properties of new chemical entities (NCE). To this end, several regulatory guidelines exist to support in vitro evaluation of NCE and decision trees that suggest when clinical evaluation is required in phase 1 evaluation of the novel drug. These clinical drug-drug interaction (DDI) studies are expensive, and thus accurate in vitro-in vivo extrapolation is crucial.
In Clinical and Translational Science, Susan E. Shoaf et al. recently described a case with tolvaptan that was flagged as a putative perpetrator of transporter DDIs using current regulatory guidelines. Two post-marketing clinical trials revealed weak but clinically irrelevant DDIs. This case highlights putative pitfalls in regulatory guidelines regarding DDIs involving breast cancer resistance protein and organic anion transporter polypeptide 1B1 and 1B3. The authors provide recommendations to improve guidelines. First, current guidelines use oral dose/250 ml to estimate intestinal concentrations of drugs without considering solubility limitations. Shoaf et al. suggest that the inclusion of solubility would improve prediction of absorption DDI, particularly for low solubility compounds. Second, current guidelines recommend fixing free fraction of drugs in plasma to 1% in cases where it is < 1%. The authors suggest that the use of precise unbound fraction of drugs might better reflect actual protein binding, free fraction, and risk of DDI. Both concordant and discordant in vitro and in vivo studies of transporter DDIs should be evaluated to understand the contribution of solubility and protein binding to these predictions. This may improve in vitro to in vivo translation for DDI studies in the future.

The comment feature is locked by administrator.