Author: Anuradha Ramamoorthy, PhD on October 18, 2022 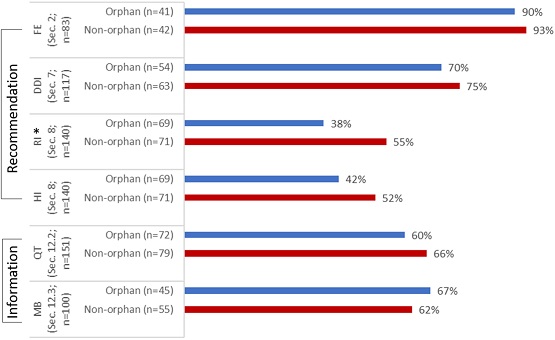
Clinical pharmacology principles are extensively leveraged during drug development, approval, and utilization to support dose optimization and therapeutic individualization, as well as to support benefit-risk assessments (particularly, in subpopulations). In fact, because of the value-add that clinical pharmacology provides, it has been referred to as the quarterback of drug development.1
During US Food and Drug Administration (FDA) review of data submitted in new drug applications (NDAs) and original biologic license applications (BLAs), information is synthesized from all relevant clinical pharmacology knowledge areas including drug disposition, biomarkers, quantitative methods, etc. to inform regulatory decisions (e.g., approvability and labeling recommendations).2 This review also identifies any critical gaps that may impact optimal therapeutic usage of the drug in patient populations that are expected to use the drug upon its approval. This information is then used to recommend studies and trials (i.e., postmarketing commitments (PMCs) and postmarketing requirements (PMRs)) that can address the knowledge gaps and can subsequently inform optimal use of the drug in clinically relevant patient subpopulations.
These review principles apply to all drugs, whether the drug is for the treatment of a common disease or a rare disease. However, it is not known if some of the clinical pharmacology studies are comparably leveraged during drug development for drugs approved to treat rare diseases or common diseases. Hsieh et al., authors of a recent paper in Clinical and Translational Science, evaluated the recommendations and information available from select clinical pharmacology studies in the labeling of new therapeutic products (drugs and biologics) that were approved between 2017 and 2019 to treat common and rare diseases (non-orphan and orphan drugs, respectively). The authors focused on recommendations and information related to food–drug interaction, drug–drug interaction (DDI), renal impairment (RI), hepatic impairment (HI), QT assessment, and human radiolabeled mass balance (MB) studies. A total of 151 new molecular entities (NMEs), including 72 orphan and 79 non-orphan drugs, were included in the analysis.
When comparing orphan and non-orphan drugs for the availability of recommendations in the labeling for effect of food in section 2, DDI in section 7, and HI in section 8, and for the availability of information in the labeling for QT assessment in subsection 12.2 and human radiolabeled MB study in subsection 12.3, overall, there were no statistically significant differences, except for renal impairment related recommendations in section 8 (see image above). The results were comparable for orphan and non-orphan drugs, but some differences were observed, for instance –
- In the case of renal impairment, although recommendations were equally available for orphan and non-orphan drugs for mild RI (98% and 100%, respectively), recommendations were less common with increasing severity of renal dysfunction for both orphan and non-orphan drugs (57% and 77%, respectively).
- Similarly, in the case of hepatic impairment, recommendations for severe HI were less common for orphan drugs when compared to non-orphan drugs (43% and 80%, respectively).
- Non-orphan drugs more frequently used TQT study design (72% vs. orphan drugs at 49%) to address questions related to cardiac safety.
- Of the 52 drugs with PMRs and PMCs, 33 orphan drugs had a combined 79 PMRs and PMCs, while 19 non-orphan drugs had 39 PMRs and PMCs established.
Overall, there appeared to be no substantial difference in how the select clinical pharmacology studies were leveraged during the development and approval of orphan and non-orphan drugs. Some information gaps related to the select clinical pharmacology studies existed for both orphan and non-orphan drugs. However, they were more pronounced for orphan drugs, resulting in more PMRs and PMCs at the time of initial drug approval, with most of the difference observed for DDI, HI, and QT assessments.
References:
1. Leinfuss, E. and Bullock, J. Clinical Pharmacology—The Quarterback of Drug Development. Clinical Researcher (2017). DOI: 10.14524/CR-17-0016
2. US Food and Drug Administration. Manual of policies and procedures. Good review practices: clinical pharmacology review of New Molecular Entity (NME) New Drug Applications (NDAs) and original biologics license applications. Center for Drug Evaluation and Research, Office of Clinical Pharmacology. MAPP 4000.4 Rev. 1. https://www.fda.gov/media/71709/download. Last accessed: August 2022.

The comment feature is locked by administrator.